Abstract
Summary: In this article we present the prototype of a bioinformatics server (Cytometry Biotechvana) for flow cytometry data analysis. The aim of this prototype is to evaluate the requirements and needs of workflows and pipelines for analysis of this type of data.
Remarks: Cytometry Biotechvana is a software application designed and developed by implementing different modules and flow cytometry packages written in R / Python / Java, and programming the different scripts and interfaces necessary to integrate the different modules as a final operational solution to a prototypical level. Through an interactive interface based on Shiny, the application allows a complete workflow, including pre-processing, data quality analysis, manual gating and six clustering algorithms (SOM, Kmeans, CLARA, Phenograph, Mclust and Hclust). The prototype has been tested against FCS files from two independent experiments to illustrate its utility, demonstrating to obtain relevant information for the final interpretation of the data. The present application serves as a starting point for future implementations, in which to integrate diagnostic algorithms or complex data visualization.
Keywords: Flow Cytometry | Bioinformatics | Machine Learning | Python | R
Sumario:En este artículo presentamos el prototipo de servidor bioinformático (Cytometry Biotechvana) para análisis de datos de citometría de flujo. A través de este prototipo pretendemos evaluar las necesidades en los flujos de trabajo analíticos y pipelines para analizar este tipo de datos.
Descripción:Cytometry Biotechvana es una aplicación software diseñada y desarrollada mediante la implementación de distintos módulos y paquetes de citometría de flujo escritos en R/Python/Java, y la programación de los distintos scripts e interfaces necesarios para integrar los distintos módulos como una solución final operativa a nivel prototípico. Mediante una interfaz interactiva basada en Shiny, la aplicación permite un flujo de trabajo completo, incluyendo preprocesado, análisis de calidad de los datos, gating manual y seis algoritmos de clustering (SOM, Kmeans, CLARA, Phenograph, Mclust y Hclust). El prototipo ha sido testado frente a FCS provenientes de dos experimentos independientes para ilustrar su utilidad, demostrando obtener información relevante para la interpretación final de los datos. La aplicación presentada sirve como punto de partida para futuras implementaciones, en las que integrar algoritmos de diagnóstico o visualización compleja de los datos.
Palabras Clave: Citometría de Flujo | Bioinformático | Aprendizaje Automático | Python | R
Disponibilidad:el código fuente se encuentra accesible en Github (URL 1)
INTRODUCCIÓN
La citometría de flujo es una técnica que posibilita el análisis multiparamétrico de células o partículas en suspensión mediante el empleo de un instrumento que recibe el nombre de citómetro [1]. Permite cuantificar de manera rápida hasta 50 parámetros diferentes de forma simultánea en millones de células por muestra [2].
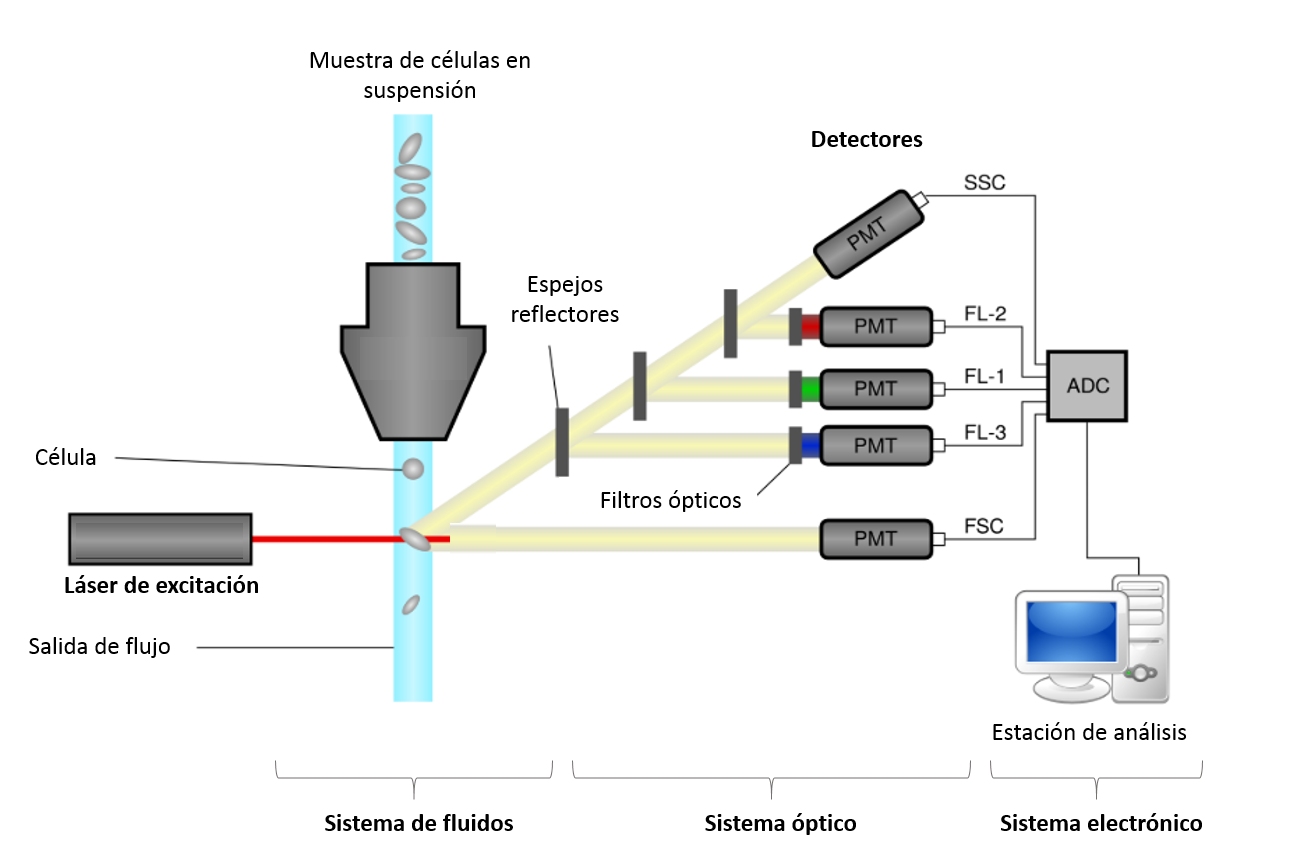
Cada célula se analiza frente a la dispersión de la luz visible y frente a diversos parámetros fluorescentes. La dispersión se mide en dos direcciones, la dispersión hacia delante o frontal (en inglés, Forward Scatter, FSC), que proporciona información sobre el tamaño relativo de la partícula, y a 90° o dispersión lateral (Side Scatter o SSC) que indica la granularidad o complejidad de la misma [1]. Por ejemplo, los granulocitos que poseen un núcleo irregular producen un mayor SSC que los linfocitos, con un núcleo más esférico [3].
Además de eso, las muestras pueden prepararse para medir su fluorescencia, ya sea propia, o mediante el uso de otros compuestos. Un reactivo fluorescente absorbe luz de colores específicos y emite luz de un color diferente, generalmente, a una longitud de onda mayor. Existen multitud de reactivos fluorescentes, como proteínas fluorescentes (Ej. Green Fluorescent Protein, GFP), anticuerpos conjugados con partículas fluorescentes (Ej. CD3 FITC) [1], usados para medir la cantidad de antígenos unidos a una superficie celular, compuestos de unión al ADN (binding dyes) para analizar multiploidías, etc. [3], de hecho en los últimos años estos han aumentado drásticamente, incrementando el número de parámetros asociados a cada análisis [1].
Aunque inicialmente los citómetros se usaban frente a células, hoy en día estos instrumentos son capaces de medir otro tipo de partículas, como pueden ser bacterias, plancton, virus, etc; por lo que se usa de manera generalizada el concepto de “evento” para cada partícula individual que el instrumento detecta. Generalmente el tamaño de las partículas analizadas se encontrará entre 1µm-30 µm de diámetro [3].
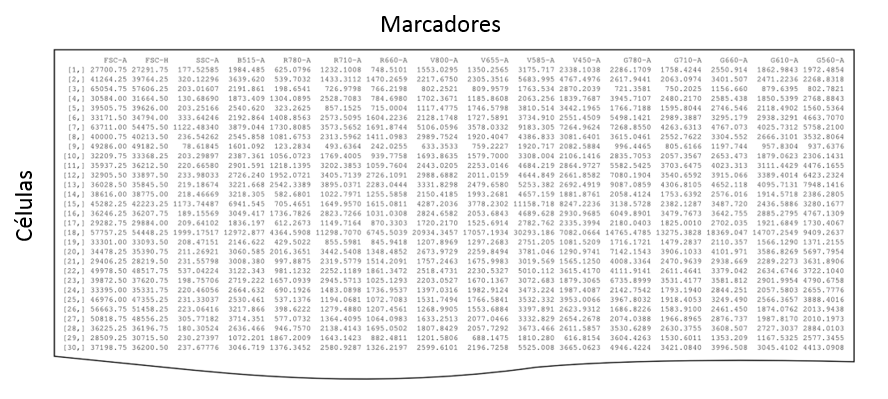
El citómetro de flujo emplea el láser como fuente de luz para producir las señales “scattered” y fluorescentes que son captadas por fotodiodos o tubos fotomultiplicadores (Fig.1). Esa señal luminosa se transforma en una señal eléctrica que finalmente será analizada y transformada por un ordenador almacenándose para su posterior análisis en un tipo de archivo binario estandarizado (FCS, Flow Cytometry Standard). El formato FCS [4] fue creado en 1984 para estandarizar los archivos de citometría de flujo, de este modo permite una mayor transferencia de resultados e interpretación de los mismos [1,5]. Los datos crudos se almacenan en forma de array, con los canales de fluorescencia representados en las columnas, y cada uno de los eventos ocupando una fila diferente (Fig. 2) [6].
La citometría de flujo es una herramienta con gran importancia y que tiene aplicación en áreas como la inmunología, la biología molecular, bacteriología, virología, biología del cáncer o la monitorización de enfermedades infecciosas [1] con ejemplos como la medición de la longitud de los telómeros o la monitorización de CD4 en VIH [6]. Por ejemplo, es especialmente útil a la hora de caracterizar células de la sangre o de la médula ósea, ya que este tipo de tejidos está compuesto por diversos tipos de células independientes, y no requieren una manipulación o tratamiento previo [3]. Además del análisis y caracterización celular, la citometría de flujo se emplea en la separación celular o “sorting”, permitiendo elegir poblaciones de interés [1].
Una de las mayores ventajas de la citometría de flujo es que es un método que permite reunir de forma muy rápida (con flujos de 500 – 5.000 partículas/s) características de células individuales. La desventaja sin embargo, es que todas aquellas células que naturalmente no se encuentren como partículas individuales, van a requerir un tratamiento previo al análisis, pudiendo alterar características de las mismas [3]. La última etapa de un análisis de citometría de flujo, y su actual cuello de botella [2], es el análisis computacional de los datos generados y la representación de los mismos [3].
En los últimos años el uso de la citometría de flujo ha aumentado considerablemente, así como el incremento del número de parámetros y la mayor complejidad de los experimentos hacen cada día más difícil la interpretación de los datos generados [1]. Aquí es donde adquiere importancia la bioinformática de citometría de flujo, el conjunto de técnicas computacionales y algoritmos de machine learning aplicados al análisis de datos generados por el citómetro [6].
Estas técnicas computacionales acompañan al usuario en el preprocesado de los datos, la identificación de las diversas poblaciones celulares y la caracterización de la información de una manera más comprensiva para su uso en diagnóstico [6]. Este esfuerzo común para el desarrollo de nuevas herramientas y métodos de análisis ha dado lugar a multitud de paquetes dentro del repositorio Bioconductor (URL 2), basado en el lenguaje de programación de R, así como el desarrollo de suites como Genepattern, que reúnen estas herramientas en un formato amigable para el usuario no experimentado con lenguajes de programación [6].
Por desgracia, la rápida evolución y desarrollo de nuevas técnicas y algoritmos, ha desencadenado que algunas de las funcionalidades de la suite de Genepattern estén obsoletas, por lo que se hace evidente la necesidad de aplicaciones que integren funcionalidades de esta, junto a nuevos paquetes incluidos en Bioconductor, para ofrecer al usuario una interfaz amistosa con la que poder analizar los datos de su experimento de citometría.
MATERIAL Y MÉTODOS
Implementación basada en shiny
Cytometry Biotechvana es una aplicación web desarrollada mediante el lenguaje de programación R y está basado en un Shiny Local Server. La aplicación se basa en un archivo principal, app.R, siguiendo la estructura de una aplicación de shiny [7], con una parte correspondiente a la ui (User interface) y otra al servidor (server). La visualización de los resultados se realiza mediante funciones básicas de R así como funcionalidades específicas de Shiny. Los scripts integrados reúnen funcionalidades que llevan desde la lectura del archivo FCS hasta el diagnóstico o interpretación de los resultados y pueden resumirse en 6 pasos principales [2,6]:
- Preprocesado de los datos (análisis de la calidad, compensación, transformación y normalización).
- Identificación de las poblaciones celulares (gating, clustering)
- Comparación entre muestras.
- Extracción de características
- Interpretación.
- Visualización
Preprocesado
Para el apartado de Preprocesado se ha utilizado módulos extraídos de la suite de Genepattern (URL 3), un repositorio web que permite el acceso a más de 180 herramientas para el análisis de expresión génica, proteómica y otros tipos de datos. Estas herramientas se encuentran disponibles en forma de módulos escritos en R, Java, Matlab, Perl o Python [8]. Los módulos utilizados y adaptados para su uso en Cytometry Biotechvana fueron PreviewFCS, CsvToFcs y FcsToCsv.
PreviewFCS y FcsToCsv son módulos alojados en el servidor de Genepattern y ejecutados mediante un script de Python. Los distintos comandos han sido modificados y adaptados para poder ejecutarlos desde la aplicación de Shiny mediante el uso del paquete reticulate [9 ] , el cual permite la interoperabilidad entre el lenguaje de R y Python.De este modo, una vez proporcionados los parámetros necesarios, la app hace un llamamiento a la suite de Genepattern, corriendo el módulo en su servidor (URL 4), y vuelca el resultado en la aplicación, permitiendo su descarga al usuario.
Por el contrario, el módulo de CsvToFcs se encontraba desactualizado en el servidor de Genepattern, por lo que se descargó la versión funcional del mismo alojada en SOURCEFORGE (URL 5). En este caso, CsvToFcs se encuentra escrito en Java, ejecutándose de forma externa a la app mediante una llamada al archivo .jar, y volcando el resultado en la aplicación de shiny.
Evaluación de la calidad
La evaluación de la calidad de los datos constituye un paso indispensable en cualquier tipo de análisis, siendo en este caso su objetivo principal detectar si las variaciones entre los distintos eventos se deben a variación de las medidas, variaciones biológicas de las muestras, o variaciones asociadas al instrumento [2], especialmente en los citómetros más modernos que manejan un gran flujo existe la necesidad de métodos de detección de errores de tipo técnico [6]. Cabe destacar que la subpoblación de células objetivo del análisis pueden suponer tan sólo un 0.05% del número total de eventos, por lo que una mala calidad de medida podría llevar a falsos positivos o a pérdida de información[10]; por este motivo, aquellas muestras con medidas alteradas por errores técnicos deberían ser eliminadas del análisis [2].
Para ello se utilizó el paquete de R flowAI [10] que evalúa tres propiedades diferentes: el caudal, la adquisición de señal y el rango dinámico [2]. El paquete de flowAI ofrece dos soluciones, una automática en forma de función, y una manual, desarrollada como aplicación de shiny. En Cytometry Biotechvana se integra esta última, adaptando el código público alojado en el repositorio de GitHub (URL 6) [10].
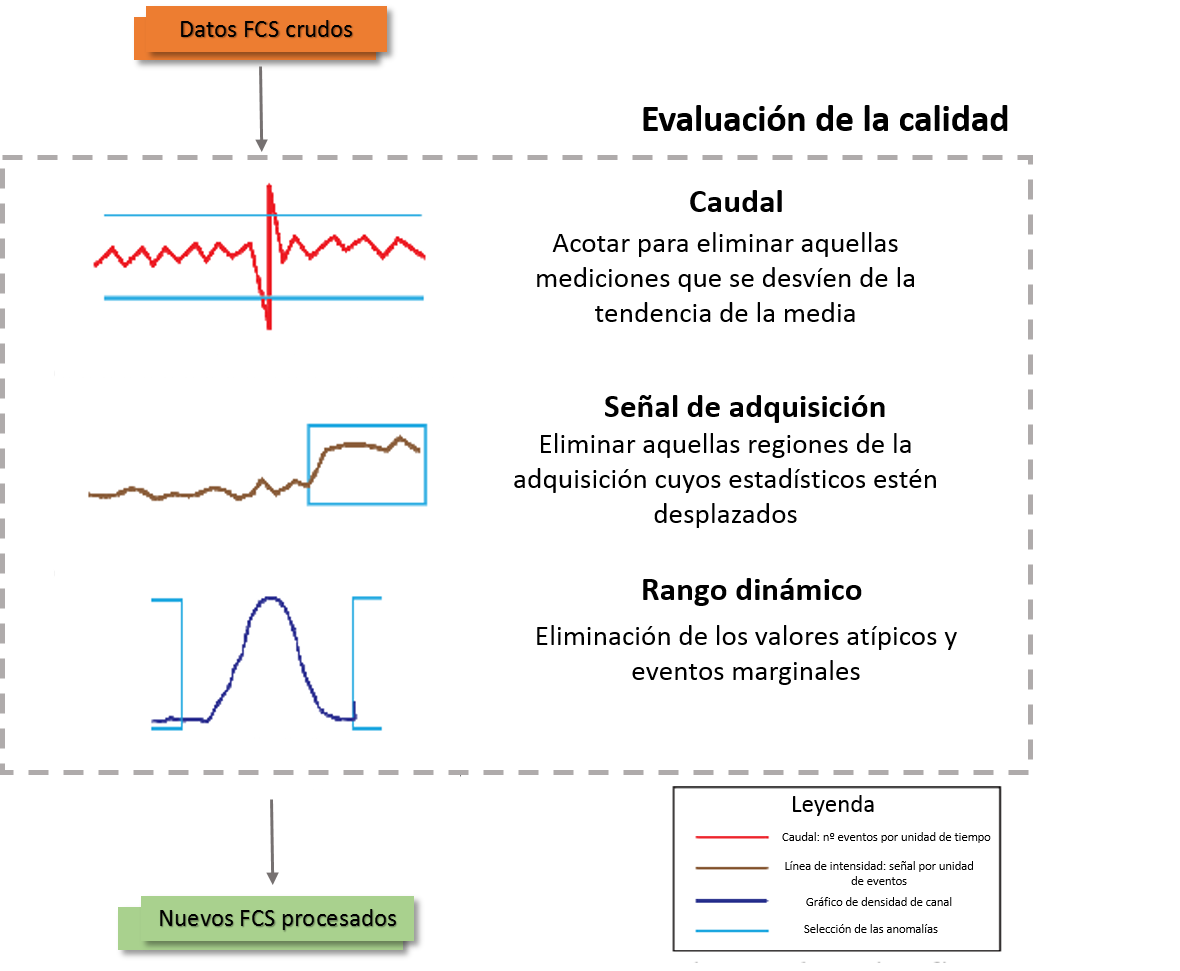
De manera interactiva el usuario puede fijar puntos de corte sobre el archivo FCS original y guardar los nuevos archivos una vez procesados, pudiendo evaluar el caudal de la medida, la adquisición de señal y el rango dinámico (Fig. 3).
En primer lugar, se evalúa la estabilidad del caudal usando una representación del número de células adquiridas por unidad de tiempo, siendo la situación óptima una gráfica con fluctuaciones no periódicas pero con una variación constante. La aparición de grandes fluctuaciones se encuentra comúnmente asociada a presencia de aire en el instrumento. En segundo lugar, se evalúa la estabilidad de la señal de adquisición a lo largo del tiempo, usando gráficos de tipo Levy-Jennings para representar la fluorescencia a lo largo del tiempo [10]. En este caso, flowAI realiza una adaptación de la función recogida en el paquete de R flowQ[11], permitiendo al usuario eliminar las regiones con una señal inestable. Se espera una distribución estable a lo largo de la medida, donde un cambio abrupto de la intensidad de la señal estaría asociado a un defecto en la detección del láser, inestabilidad del voltaje o una mala preparación de la muestra [10].
El último paso de la evaluación de la calidad consiste en acotar correctamente el límite superior e inferior del rango dinámico. Por limitaciones del rango dinámico del citómetro utilizado, todas aquellas medidas cuyo valor real caiga por encima de este, se acumularán en el último canal, recibiendo comúnmente el nombre de valores marginales. El caso del límite inferior es algo diferente. Todas las señales provenientes de los canales de dispersión (FSC/SSC) que muestran un valor por debajo de 0 son eliminados; no obstante, en los canales de fluorescencia se aceptan fluctuaciones de la señal cercanas a 0 (auto fluorescencia, superposición de espectros…etc). En estos casos el problema aparece cuando por culpa de una inestabilidad en el voltaje, cambios en el caudal, etc, esos valores negativos pueden verse intensificados, repercutiendo de manera negativa en pasos posteriores del análisis[10].
Compensación, Transformación y Normalización de los datos
De manera previa a otros pasos del análisis de datos de citometría de flujo, existe la necesidad de preparar o procesar los datos crudos para corregir diversos errores asociados y evitar introducir sesgos en posteriores pasos del proceso.
Cuando se utiliza más de un fluorocromo con el mismo láser, es común que los espectros de emisión se superpongan, un fenómeno que recibe comúnmente el nombre de “spillover”. Esta superposición se corrige mediante el empleo de una serie de muestras representativas, cada una marcada con un fluorocromo diferente. La señal que debe eliminarse de cada canal se computa mediante una serie de ecuaciones lineales sobre estas muestras, produciendo la llamada matriz de spillover. Finalmente, esta matriz se invierte y multiplica sobre los datos crudos del citómetro, proceso que recibe el nombre de compensación, corrigiendo el problema inicial [6].
Normalmente, los citómetros actuales registran los valores de forma lineal, por lo que es común incluir un paso de transformación logarítmica de los valores de manera previa al análisis; además, es común encontrar valores negativos derivados de la compensación de los datos y que serán malamente manejados por la transformación logarítmica, por lo que actualmente, también se están desarrollando otros tipos de transformación como la Logicle o Hyperlog [6].
Adicionalmente, de manera previa al paso de clusterización, es importante normalizar los datos de entrada, especialmente en experimentos multicentro, para eliminar la variabilidad técnica asociada [2,6]
Este conjunto de pasos para la preparación de los datos de citometría se realiza mediante CytoTree [5], un paquete escrito en R y obtenido del repositorio de Bioconductor. Cytotree cuenta con funciones integradas como CytoTree::runExprsExtract que permiten de manera simultánea compensar y transformar un fichero FCS. En Cytometry Biotechvana, el usuario puede compensar sus datos bien a través de una matriz de compensación facilitada externamente o bien integrando la propia matriz dentro del fichero FCS. Inicialmente no se aplica una transformación de los datos, aunque para trabajar con los sucesivos pasos de clustering, CytoTree crea un tipo de objeto CYT mediante la función createCYT, normalizando simultáneamente los datos logarítmicamente.
Gating
El paso más crítico y que más tiempo conlleva en cuanto al análisis manual de los datos, es la identificación de poblaciones celulares homogéneas dentro del conjunto de eventos, un proceso que comúnmente recibe el nombre de gating [2]. Las técnicas de gating suelen dividirse en dos categorías principales, gating secuencial o manual, comúnmente referido a él como “gating”, y el “gating” automático [2].
El gating manual consiste en dibujar una región alrededor de un grupo de células para poder aplicar a esa región otros parámetros de interés (ej. otros marcadores) [1]. Esto permitía el análisis de sub-sets de células dentro de una población y evaluarlas de forma multiparamétrica [3].
De este modo los datos del fichero FCS pueden representarse en gráficos de dos dimensiones, “scatter plot”, permitiendo una separación de un subset de estos datos basándose en la intensidad de fluorescencia, los cuales reciben el nombre de “gate” [6].
En la aplicación el usuario puede establecer unos límites de corte para seleccionar subpoblaciones del total de datos, y después representar dichos datos en un gráfico 2D seleccionando que marcadores quiere ver representados en los ejes.
La funcionalidad de Gating se implementa mediante la función “gatingMatrix” de CytoTree [5], que permite realizar un gating manual sobre los datos una vez compensados proporcionando límites superiores e inferiores para los canales FSC/SSC. Para complementar este apartado se incluye una representación del subset de células en un gráfico 2D con los diferentes marcadores en los ejes, desarrollado mediante el paquete Plotly [7].
La principal problemática de este tipo de análisis es que depende más de la experiencia y conocimientos del usuario (subjetividad), y no tanto de un método estandarizado. Además, pasa por alto datos con grandes dimensiones que no pueden ser representados en gráficos 2D, perdiendo información multidimensional [2], la variabilidad entre muestras, su empleo con grandes matrices de datos puede ser muy costosa en tiempo, etc. Todo esto mejoraría con el empleo del gating automático, mediante diversos algoritmos que realizarían esa separación de diversas poblaciones computacionalmente; no obstante, el gating de manera manual se sigue manteniendo como la mejor solución para identificación de poblaciones raras con una mala separación del resto de eventos [6].
Clustering
Por otro lado existen algoritmos que se encargan de realizar el gating de manera automática sobre el conjunto de poblaciones presentes en el fichero FCS, un área de investigación que ha crecido de manera considerable desde 2008 [6]. Aunque también se debe tener en cuenta que automatizar el paso de gating puede llegar a resultar complicado porque las poblaciones son asimétricas o solapan, existen eventos “outliers”, errores en los canales de fluorescencia, etc. Todo esto unido a otros factores puede introducir sesgos o errores que serán arrastrados a otros puntos del análisis y su posterior interpretación [2.
El gating automático puede dividirse en dos tipos:
- Métodos supervisados
- Métodos no supervisados
Métodos supervisados
La identificación celular basada en métodos supervisados requiere que los datos empleados por el usuario contengan por un lado las medidas para cada uno de los eventos, y por otro lado, una etiqueta o definición asociada a cada uno de los eventos. Es decir, esa marca señalará a qué clase pertenece cada célula o evento, y por tanto todos aquellos eventos asociados a una misma clase constituirán una población celular [2]
Métodos no supervisados
En el caso de los métodos no supervisados, el usuario no tiene ninguna clase definida como referencia, es decir, no existe una variable dependiente. Dentro de los métodos no supervisados, los más utilizados en la citometría de flujo son los algoritmos de clustering, un tipo de algoritmos que se encargan de identificar aquellas células/eventos que se encuentran dentro de un mismo clúster [2], es decir, células que presentan un perfil de expresión similar . Además, en contraste con los anteriores, resultan especialmente útiles frente a análisis exploratorios, por ejemplo, para investigar la diversidad celular de una muestra[12].
El clustering trabaja de forma multidimensional, superando las limitaciones que encontradas con el gating manual. Una vez preparadas las muestras, la estrategia para determinar los distintos clúster va a depender del tipo de algoritmo utilizado [2], teniendo en cuenta, entre otros, factores como la eficiencia computacional y de clusterización, ya que estos van a depender de las propiedades matemáticas del algoritmo [12] . Además, la mayoría de estos algoritmos requieren una etapa previa de reducción de datos o downsampling para disminuir la complejidad del análisis [1].
Existe una gran variedad de algoritmos de clustering disponibles, en forma de paquetes de R, integrados en diversas herramientas, o como aplicaciones independientes. L.Weber y M.Robinson [12] realizaron una comparación de 18 algoritmos distribuidos de manera gratuita, comparando su eficacia a la hora de detectar diferentes poblaciones celulares, observando uno de los resultados más precisos y rápido es con el algoritmo SOM (Self-organizing maps). Este algoritmo resulta ideal para realizar análisis exploratorios rápidos en grandes conjuntos de datos debido a su rapidez de procesamiento. No obstante, para obtener un rendimiento óptimo, es importante que el usuario seleccione el número adecuado de clústeres que deben ser predichos [2].
En Cytometry Biotechvana, al igual que en el apartado de gating, los datos crudos del FCS son compensados mediante CytoTree [5], y a diferencia de este, son también normalizados logarítmicamente, obteniendo un objeto de tipo “CYT”.
El apartado de clustering integra el uso de 6 algoritmos no supervisados a través del paquete de Cytotree [5] sobre el objeto CYT generado anteriormente: self-organizing maps (SOM), k-means clustering (Kmeans), clustering large applications (Clara), Phenograph, hierarchical clustering (hclust) y mclust. Si además el número de eventos totales es superior a 100.000 la app permite usar la opción de reducción de tamaño muestral para reducir el tiempo de computación, especialmente con mclust y hclust, no recomendados para FCS de gran tamaño.
Tras el clustering, se aplica a cada clúster una reducción de dimensionalidad con los algoritmos: PCA (análisis de componentes principales), tSNE (t-Distributed Stochastic Neighbor Embedding), UMAP (uniform manifold approximation and projection) y mapas de difusión [5]. Este tipo de algoritmos permiten reducir matrices de datos de gran dimensión, creando subsets de 2 o 3 dimensiones que puedan ser fácilmente representados en gráficos 2D-3D [2]
Finalmente la visualización de los clúster se desarrolló mediante la integración de los datos generados por Cytotree [5] y el paquete de creación de gráficos y elementos visuales de Plotly [7] representando los datos en un gráfico de 2 dimensiones.
Material de training
Para el desarrollo y testeo de las distintas funcionalidades de Cytometry Biotechvana se usaron dos archivos FCS alojados en el repositorio público de FlowRepository (FlowRepository.org), una página web creada para aumentar la transparencia y accesibilidad a análisis, protocolos y datos provenientes de citometría de flujo [13]. El primero de ellos “Pre RNA Protein staining_CD4,2f,14_001.fcs”, alojado bajo el identificador FR-FCM-ZZC7, corresponde con datos provenientes de un experimento de citometría de flujo de RNA [14].
El segundo dataset utilizado es “Pronk CytometryA Figure2.fcs”, incluido en el experimento “FR-FCM-ZZ6L”. Se trata de un FCS de células de médula ósea de un individuo sano, usado en un manuscrito sobre el análisis de subsets de células raras [15], y bajo diversas modificaciones, para el testeo de diversos algoritmos de clustering [12].
RESULTADOS
Implementación Back-end
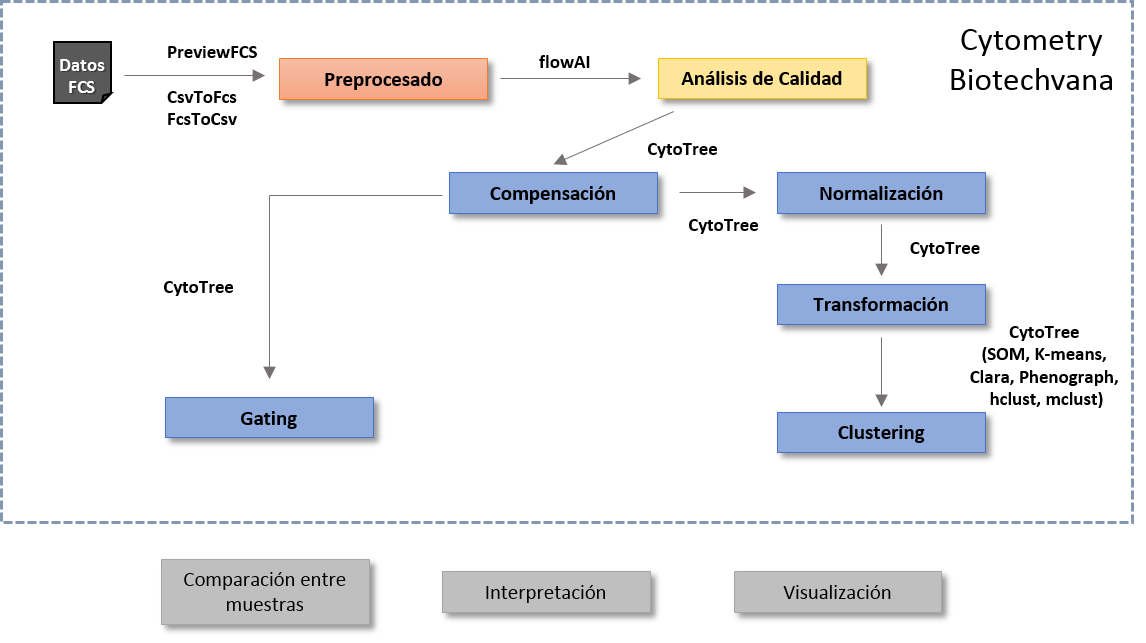
Cytometry Biotechvana implementa un pipeline dirigido al análisis de datos de citometría de flujo que se divide en tres bloques principales, ejemplificados con diferente color en Fig. 4.
En primer lugar la aplicación toma como fichero de entrada un archivo FCS, y extrae un resumen de la información contenida en este, mediante el módulo PreviewFCS. Este módulo junto a los módulos de CsvToFcs y FcsToCsv, que permiten la interconexión de formato entre archivos FCS-CSV, constituirían el bloque de preprocesado del pipeline.
El segundo bloque se compondría del apartado de calidad, con el uso del módulo de flowAI. Este apartado permite evaluar la calidad (en bases a parámetros técnicos) del fichero FCS de entrada, modificar el número de eventos presente en este, y generar un fichero FCS nuevo que será utilizado en posteriores pasos del proceso.
El último y más extenso bloque consistiría en el análisis de los datos de citometría de flujo. Este se divide en dos ramas principales, correspondientes por un lado al análisis de gating, y por otro lado al clustering, ambas basadas en el paquete de Cytotree [5]. Sendas ramas pasan por un nodo común, consistente en la compensación de los datos FCS; además, en el caso del clustering, también es necesario realizar un paso de normalización y transformación de los datos, antes de aplicar los diversos algoritmos de clustering.
El flujo de trabajo recomendado para el análisis de datos provenientes de citometría de flujo es el ejemplificado en la Fig. 4 no obstante, la mayoría de sub-bloques pueden accederse y ejecutarse de manera independiente, salvo a excepción de la compensación, normalización y transformación, que se encontrarán ligados al gating o clustering.
Idealmente, aunque estos representarían futuros desarrollos de la aplicación, el pipeline proseguiría tomando el resultado de gating o clustering, aplicando algoritmos de comparación de muestras, interpretación de resultados, u otros métodos de visualización y representación de los mismos.
Implementación Front-end
La interfaz visual de la aplicación de Cytometry Biotechvana está implementada mediante la herramienta Shiny [7], lo que permite obtener una app de tipo web, sencilla, interactiva y reactiva.
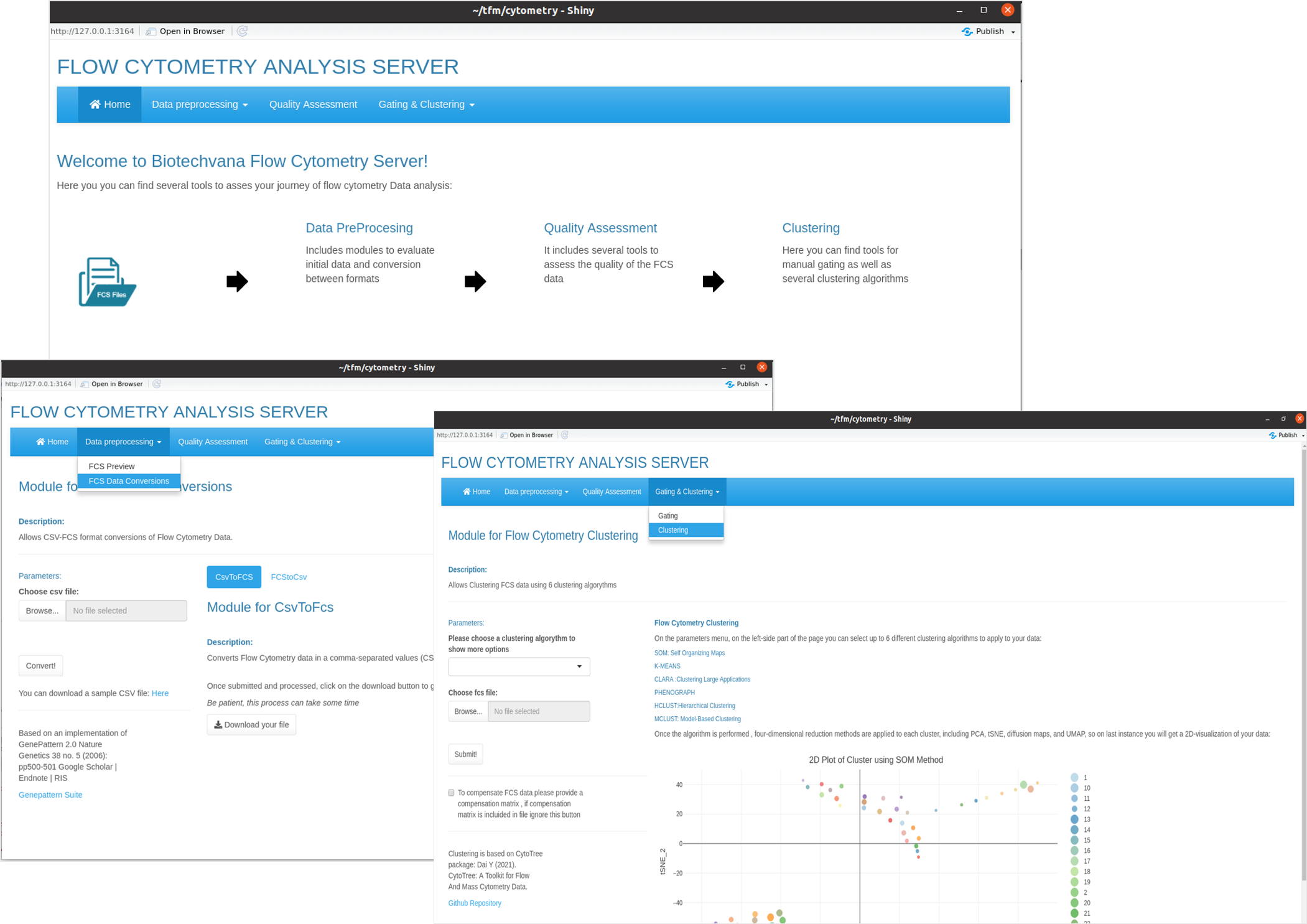
El prototipo de la app se divide en un menú principal en el que destaca la página de inicio, y los tres bloques de análisis comentados anteriormente: preprocesado, análisis de calidad y gating/clustering. Además, tanto el apartado de preprocesado como el de gating/clustering dan lugar a un submenú desplegable (Fig. 5).
El submenú del apartado de preprocesado, por un lado da acceso a las funcionalidades de conversión de archivos CSV (Comma-separated-values) al estándar de citometría FCS y viceversa englobados en “FCS Data conversions”, y por otro, al apartado de FCS Preview, que permite obtener información inicial sobre el archivo de análisis.
Esta funcionalidad de la app puede ser de vital importancia para el usuario no familiarizado con la cantidad de detalles sobre los datos que pueden almacenarse, visualizando en forma de listado los metadatos del archivo, entre los que se encuentran el número de eventos contabilizados o número de parámetros incluidos en el experimento [8] con sus respectivas medias, valores mínimos-máximos, etc, así como las keywords registradas, versión del archivo FCS o fecha de creación del mismo.
En el caso de la funcionalidad de conversión de archivos, su valor destaca especialmente si en posteriores o complementarios análisis que el usuario necesita realizar, utiliza herramientas que puedan trabajar con matrices simples, pero no puedan ser capaces de analizar un archivo FCS [8].
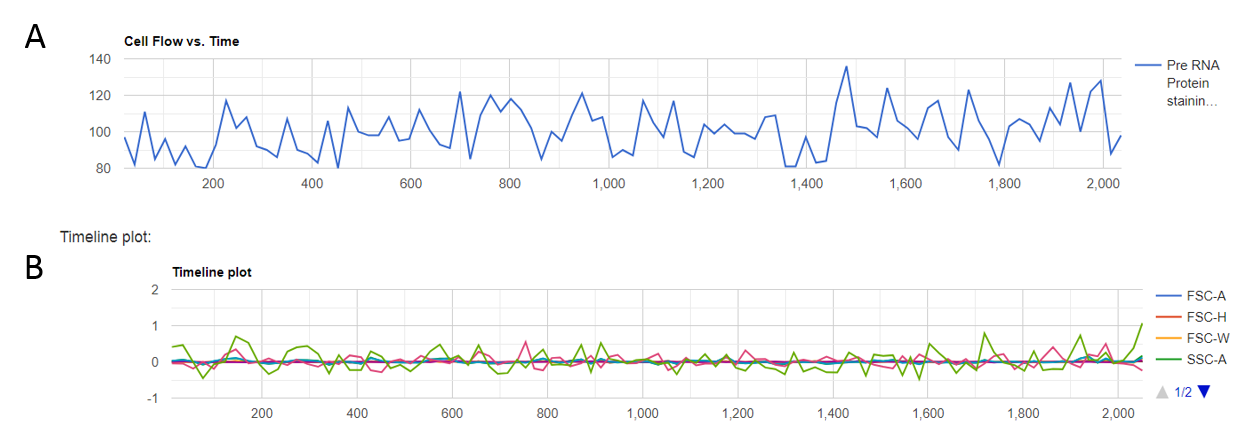
El segundo apartado, el de análisis de calidad, permite cargar un archivo FCS y visualizar distintas gráficas interactivas agrupadas en diversas pestañas en el margen derecho de la interfaz (Cell numbers, Time flow, Timeline, Margin Events, QA score, Summary). En base a los parámetros representados y la interpretación del usuario, este dispone de una barra interactiva en el margen izquierdo que le permite “recortar” los eventos o registros del archivo FCS en función del tiempo, generando y descargando nuevos FCS procesados, para posteriores pasos del análisis.
En el último submenú de la web se encuentra gating y clustering. El primero está compuesto por una página única que permite cargar el archivo FCS en el margen izquierdo, con el parámetro opcional de añadir una matriz de compensación; mientras que en el margen derecho se representan los diversos gráficos una vez lanzado el análisis: gráfico FSC/SSC de los datos crudos antes de realizar gating, mismo gráfico pero una vez han sido aplicados los límites de corte para el gating, y en último lugar, un gráfico interactivo en el que el usuario puede seleccionar qué canales quiere ver representados en cada uno de los ejes.
Por último, la interfaz del apartado de clustering es muy similar a la de gating, con la diferencia de que en este caso el usuario tiene un desplegable al margen izquierdo en el que seleccionar el tipo de gating: SOM, Kmeans, CLARA, Phenograph, Mclust, y Hclust. En función del algoritmo seleccionado, el aspecto de la interfaz variará, mostrando un apartado de parámetros que resultan específicos para cada algoritmo. El usuario puede así definir por ejemplo el número de clúster, porcentaje de reducción muestral, etc. Una vez definido el valor de estos y enviar la orden a través del botón de “Submit”, se representará un gráfico 2D con los clústeres identificados por el algoritmo seleccionado.
Los gráficos generados tanto en el apartado de gating como en el apartado de clustering pueden descargarse en formato “.png” para su posterior utilización.
Código fuente y ejecutables
El código de la aplicación se encuentra disponible en el repositorio público de GitHub (URL 1). Al tratarse de una aplicación basada en Shiny, esta puede ser alojada en un servidor, o correrse de manera local. Para esto último es necesario disponer del lenguaje de programación R instalado en el sistema, ya que este actuará como “back-end” de la aplicación, mientras que el navegador web hará de front-end. Para ejecutar la aplicación basta con ejecutar desde R el fichero “cytometry_app.R”. Es importante seguir la jerarquía de archivos que se muestra en GitHub, por lo que se recomienda crear una carpeta en la que irá el fichero principal de la APP “cytometry_app.R”, el fichero con funciones auxiliares “global.R”, el ejecutable de Java “csv2fcs”, y las carpetas de “Python” y “www”. Para su correcto funcionamiento, también será necesario disponer de Python y Java instalados en el sistema.
La aplicación está desarrollada para que todos los paquetes auxiliares necesarios para su correcto funcionamiento sean descargados de forma automática, no obstante en el repositorio de GitHub puede encontrarse más información sobre la versión de R utilizada y algunas notas auxiliares ante posibles problemas de instalación.
Prueba de concepto
Durante el desarrollo del prototipo se usaron los archivos “Pre RNA Protein staining_CD4,2f,14_001.fcs” y “Pronk CytometryA Figure2.fcs” para testear las diversas funciones y flujos de trabajo, mostrando a continuación un ejemplo de evaluación de calidad, y de identificación y diferenciación de poblaciones celulares desarrollado en Cytometry Biotechvana.
Prueba de concepto 1. Evaluación de la calidad de un archivo FCS
Se evaluó la calidad del archivo “Pre RNA Protein staining_CD4,2f,14_001.fcs”, un conjunto de células mononucleares de sangre periférica humana (PBMC) marcadas fluorescentemente con marcaje anti-CD4 [14]. Al cargar el archivo en el apartado de calidad de la app se observó que este contaba con 9 canales, uno de tiempo, 6 correspondientes a la dispersión de la luz, y 2 de marcadores (PE-A y HV 450-A); y que tenía un número total de 10.060 células.
El flujo de células registrado por el instrumento a lo largo del tiempo presentaba variaciones esperadas, sin cambios abruptos que indicaran una posible presencia de burbujas o agrupaciones de células como se muestra en laFig. 6 A. La señal de adquisición registrada por el instrumento se evalúa en el apartado de Timeline, dentro del cual se obtiene un gráfico que representa las diversas mediciones registradas por el láser a lo largo del tiempo para cada uno de los canales como se observa en la Fig. 6 B . Las variaciones obtenidas, al igual que en el caso anterior, entraban dentro de los valores esperados, sin desviaciones pronunciadas, destacando quizá, una mayor separación en la parte final del mismo, que podría solucionarse descartando del FCS los eventos registrados en el último rango de tiempo. Por último, la cantidad de eventos marginales registrados era casi nula, por lo que teniendo en cuenta estos parámetros, eliminando las últimas células medidas por el instrumento, el nuevo fichero FCS estaría preparado para posteriores análisis.
Prueba de concepto 2. Identificación y diferenciación de poblaciones celulares presentes en un fichero FCS
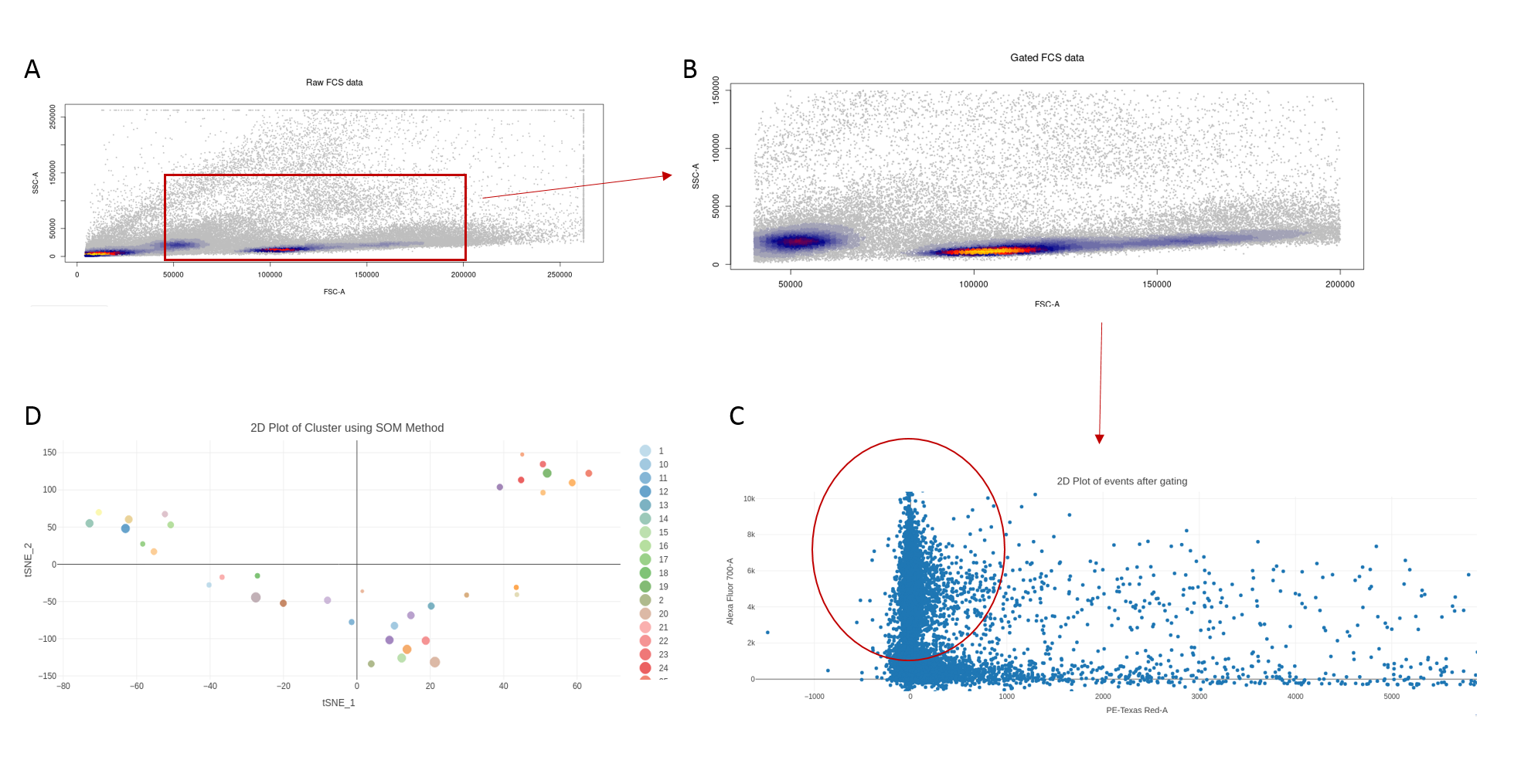
En esta segunda aproximación se utilizó el fichero “Pronk CytometryA Figure2.fcs” proveniente de una muestra de células de médula ósea humana [15]. En primer lugar, usando el módulo de preview FCS dentro del menú de preprocesado, se obtuvo una primera aproximación del conjunto de datos recogidos en el archivo. El FCS recogía información de 119.959 células y 14 canales, informando además del target conjugado a los distintos fluoróforos (Por ejemplo Alexa flúor 700-A con CD45, o PE-Texas Red-A con PI (Yoduro de Propidio). Dentro de la información proyectada, podía observarse también la presencia y valores de la matriz de compensación incluida dentro del propio FCS, por lo que en posteriores apartados no sería necesario proporcionarla.
Usando como guía el estudio de Nilsson et al [15], se aplicaron diversos parámetros de gating para la identificación de alguna de las subpoblaciones celulares en la muestra. En primer lugar, en el apartado de gating, tras compensar los datos, se obtuvo una representación inicial de las células (Fig. 7 A ), mostrando los diferentes eventos en un gráfico de densidad en base a los canales FSC/SSC. Pese a encontrar células dispersas a lo largo de todo el gráfico, estas se agrupaban principalmente en torno a 3 núcleos o zonas con una mayor densidad, de tal modo que aplicando el gating señalado, seleccionaríamos los dos núcleos recogidos en la parte central del gráfico (Fig. 7 B ). Finalmente, se representó el conjunto de células seleccionadas en base a los canales Alexa flúor 700-A (CD45) y PE-Texas Red-A (PI) (Fig. 7). El yoduro de propidio se usa principalmente para el estudio de apoptosis, ya que este no penetra de manera normal en las células a no ser que su membrana se encuentre dañada [16], por lo que el subconjunto de células de interés deberían encontrarse lo más cercanas al 0 teniendo en cuenta el eje X. Por otro lado, CD45 es un marcador expresado en células hematopoyéticas [17], por lo que respecto al eje Y, la región de células de interés sería aquella con los valores más alejados del origen de coordenadas. Cabe destacar que en este caso, la subpoblación de células de interés (Fig. 7 C), constituiría un bajo porcentaje respecto al conjunto de eventos totales registrados por el citómetro. Posteriores pasos de gating no recogidos en este prototipo permitirían identificar las diferentes subpoblaciones dentro del conjunto de células hematopoyéticas, de la misma manera que se describe en el experimento original [15].
En último lugar, se testearon los diferentes algoritmos de clustering usando como valor inicial de número de clúster, 40. Al igual que en estudios previos donde se utiliza este conjunto de datos [12], se observó que el algoritmo que ofrecía un mejor resultado y un menor tiempo de respuesta era SOM (self-organizing map) (Fig. 7 D) con un tiempo de ejecución de alrededor de 1 minuto, frente a KMeans (18 minutos) o CLARA (30 minutos). El resto de algoritmos no se testearon para este caso al recomendarse su uso con un conjunto de datos de menor tamaño.
Además de los resultados comentados, los tiempos de procesamiento para los diversos análisis fueron inferiores con el archivo “Pre RNA Protein staining_CD4,2f,14_001.fcs”. Esa velocidad de procesamiento correlaciona con el tamaño del archivo utilizado, número de eventos y canales de fluorescencia presentes en este, recomendando tener este factor altamente en consideración a la hora de procesar archivos de citometría de flujo usando el presente prototipo de la app.
DISCUSIÓN
La citometría de flujo es una técnica ampliamente utilizada en laboratorios de investigación y diagnóstico, con experimentos que producen resultados complejos con mucha información relevante para entender problemas biomédicos. El principal problema sin embargo, se encuentra en poder aprovechar toda esa información dada la dificultad de análisis [2].La evaluación manual de los datos puede resultar muy laboriosa y requerir mucho tiempo, mientras que los algoritmos de machine learning son capaces de superar esas limitaciones. En los últimos años este tipo de algoritmos están adquiriendo gran importancia, a la vez que revolucionan la investigación en el mundo de la salud [2], por lo que usar interfaces sencillas que los integren puede ser de gran relevancia para su uso en diagnóstico o investigación, por un personal sin habilidades de programación.
Cytometry Biotechvana es un prototipo de aplicación que marca la estrategia a seguir para el análisis de datos procedentes de citometría de flujo, usando una interfaz sencilla y amigable. Esta contiene los elementos básicos para visualizar propiedades de un archivo FCS, analizar la calidad de los datos de partida, aplicar un gating manual inicial o testear diferentes algoritmos de clustering.
Cada una de las funcionalidades presentadas proporciona el punto de partida para futuros desarrollos o implementaciones. Destacando, por ejemplo, la posibilidad de poder aplicar un gating manual secuencial, ya que en el presente prototipo sólo puede aplicarse una única vez; la posibilidad de permitir definir un mayor número de parámetros al usuario a la hora de realizar cada uno de los análisis, como incluir otros métodos de transformación de los datos (transformación lógica o “arcsinh”) o procesar varios archivos FCS de manera simultánea.
Además de las mejoras comentadas, una futura herramienta completamente funcional debería incluir nuevos algoritmos o módulos, capaces de completar aquellos pasos finales del proceso que permitan la comparación entre diversos experimentos, visualizaciones más complejas de los datos, o incluso algoritmos predictivos que ayuden en el proceso de diagnóstico.
INSTALACIÓN
El prototipo de Cytometry Biotechvana puede ejecutarse localmente en cualquier ordenador que cuente con los lenguajes de programación de R, Python y Java. Esta herramienta se distribuye a través del repositorio de Github (URL 1) con todos los scripts y materiales necesarios para su correcta ejecución. Una vez descargado y respetando siempre la jerarquía de directorios, se ejecutará desde R haciendo un llamamiento al archivo de “cytometry_app.R”, mediante el comando runApp(“cytometry_app.R”) o usando la IDE de RStudio (URL 7). El script del prototipo está diseñado para que todas las dependencias y paquetes adicionales sean descargados de manera automática al ejecutar la aplicación por primera vez.
REQUERIMIENTOS
Cytometry Biotechvana requiere tener R (URL 8), Python (URL 9) y Java (URL 10) instalados en el sistema. El prototipo ha sido desarrollado mediante la versión de R 4.0.5, Python 3.8.10 y Java (JDK) 11.0.11, por lo que se recomienda su uso para evitar problemas de incompatibilidad de paquetes. Debido a la complejidad y tiempo de procesamiento de algunos de los algoritmos incluidos en el prototipo, se recomienda usar un ordenador con un procesador i5-4210U 1.70GHz o superior, y una memoria RAM de mínimo 4GB.
CONCLUSIONES
Del presente desarrollo realizado sobre la implementación de un servidor bioinformático que permita analizar datos provenientes de citometría de flujo, se pueden deducir las siguientes conclusiones:
- La herramienta, Cytometry Biotechvana, ha sido desarrollada con éxito, ejerciendo su función de prototipo, evidenciando las funcionalidades necesarias de un flujo de análisis de datos procedentes de citometría de flujo.
- La aplicación posee una interfaz sencilla e intuitiva, permitiendo integrar algoritmos complejos al alcance del usuario no experimentado en programación y mostrando su utilidad frente a datos reales provenientes de dos experimentos independientes.
- El presente prototipo puede ser utilizado como punto de partida para abordar futuras mejoras y desarrollos, convirtiéndose en una aplicación cerca de mercado; destacando la inclusión de un mayor número de parámetros que sean modificables por el usuario, o la implementación de algoritmos predictivos de diagnóstico.
AGRADECIMIENTOS
El presente trabajo y prototipo de Cytometry Biotechvana ha sido realizado bajo el marco del trabajo de fin de máster (TFM) del máster de Análisis Bioinformático Avanzado de la Universidad Pablo de Olavide (Sevilla).
LICENCIA Y DISTRIBUCIÓN
Cytometry Biotechvana es un prototipo de libre acceso distribuido bajo una licencia GNU GPLv3 (URL 11).
REFERENCIAS
- McKinnon KM: Flow Cytometry: An Overview. Curr Protoc Immunol 2018, 120:5.1.1–5.1.11.
- Montante S, Brinkman RR: Flow cytometry data analysis: Recent tools and algorithms. Int J Lab Hematol 2019, 41:56–62.
- Givan AL : Flow Cytometry: An Introduction. Methods Mol.Biol 2011, 699:1–29.
- Murphy RF, Chused TM: A proposal for a flow cytometric data file standard. Cytometry 1984, 5:553–5 .
- Dai Y, Xu A, Li J, Wu L, Yu S, Chen J, et al. CytoTree: an R/Bioconductor package for analysis and visualization of flow and mass cytometry data. BMC Bioinformatics 2021, 22:1–20 .
- O’Neill K, Aghaeepour N, Špidlen J, Brinkman R. Flow Cytometry Bioinformatics. PLoS Comput Biol 2013, 9:e1003365.
- Sievert C. Interactive Web-Based Data Visualization with R, plotly, and shiny (1st ed.) Chapman and Hall/CRC, 2020 .
- Spidlen J, Barsky A, Breuer K, Carr P, Nazaire MD, Hill BA, et al. GenePattern flow cytometry suite. Source Code Biol Med 2013, 8:14.
- JJ Allaire, Ushey K, Tang Y, Eddelbuettel D. reticulate: R Interface to Python 2017.
- Monaco G, Chen H, Poidinger M, Chen J, De Magalhães JP, Larbi A. FlowAI: Automatic and interactive anomaly discerning tools for flow cytometry data. Bioinformatics 2016, 32:2473–80.
- Gentleman R, Hahne F, Kettman JR, Le Meur N GN. R/bioconductor package flowQ: Quality control for flow cytometry 2018 .
- Weber LM, Robinson MD. Comparison of clustering methods for high-dimensional single-cell flow and mass cytometry data. Cytom Part A 2016, 89:1084–96.
- Spidlen J, Breuer K, Rosenberg C, Kotecha N, Brinkman RR. FlowRepository: A resource of annotated flow cytometry datasets associated with peer-reviewed publications. Cytom Part A 2012, 81 A:727–31.
- . Van Hoof D, Lomas W, Hanley MB, Park E. Simultaneous flow cytometric analysis of IFN-γ and CD4 mRNA and protein expression kinetics in human peripheral blood mononuclear cells during activation. Cytom Part A 2014, 85:894–900.
- Rundberg Nilsson A, Bryder D, Pronk CJH. Frequency determination of rare populations by flow cytometry: A hematopoietic stem cell perspective. Cytom Part A 2013, 83:721–7.
- Riccardi C, Nicoletti I. Analysis of apoptosis by propidium iodide staining and flow cytometry. Nat Protoc 2006, 1:1458–61.
- Woudstra L, Biesbroek PS, Emmens RW, Heymans S, Juffermans LJ, van der Wal AC, et al. CD45 is a more sensitive marker than CD3 to diagnose lymphocytic myocarditis in the endomyocardium. Hum Pathol 2017, 62:83–90.
URLs
- Repositorio de Cytometry-Biotechvana:https://github.com/aligogon/Cytometry-Biotechvana
- Bioconductor:http://bioconductor.org
- Genepattern:http://www.genepattern.org
- Servidor Genepattern:http://genepattern.broad institute.org/gp/
- Repositorio CsvtoFcs:https://sourceforge.net/projects/flowcyt/files/FCS/FCS%20Utilities/
- flowiQC_shinyAPP:https://github.com/SIgNBioinfo/flowiQC_shinyAPP
- RStudio:https://www.rstudio.com
- R:https://cran.r-project.org/
- Python:https://www.python.org/downloads/
- Java:https://www.oracle.com/java/technologies/downloads/
- GNU General Public License:https://www.gnu.org/licenses/gpl-3.0.html





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}