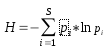
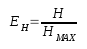


Abstract
Summary: In this paper we introduce version 2.0 of CheckAlign, an open source application oriented to bioinformatic analyses based on information theory and the Shannon-Weaver algorithm.
Remarks: The update consists in the following: (a) a comprehensive source code optimization to adapt CheckAlign to the new versions and updates of the most common operating systems; (b) the implementation of an additional utility for estimating diversity index measures for one or more biological or molecular samples based on the proportional abundance of species in the samples.
Availability: CheckAlign 2.0 is an open source software project available both as an online server and as standalone software. The server is publicly accessible at [URL1]. Software and source code can be downloaded from [URL2].
OVERVIEW
Information theory is a branch of applied mathematics involving the quantification of information. Soon after its inception, information theory [1] was applied to distinct areas of biological research. Of significant interest was the sequence logo methodology [2,3], which offers graphical representations of DNA or protein multiple alignments, providing a statistically relevant visualization of the consensus of a set of sequences, their common information content, and the frequency of all possible DNA and amino acid states per alignment position. In 2008, taking this methodology into consideration, we designed the first version of CheckAlign, a logo-maker application that builds sequence logo representations online and on PCs. In this paper, we present an update of CheckAlign to version 2.0. The update is based on a source code optimization to adapt the logo maker to the updates of the most common operating systems and an implementation of the Shannon-Weaver algorithm [1] to characterize species diversity in a community. This means that CheckAlign now focuses not only on alignment analyses, but also on other ecological or molecular analyses concerning the rarity and commonness of species in a community. The ability to quantify diversity in this manner is useful for understanding the structure of a biological community. This is valid for evaluating the diversity of an ecological community of organisms but also for determining the diversity, for instance, of a molecular population of mobile genetic element lineages in a host genome. In other words, the concept of community can be considered at both ecological and molecular levels. Taking this into account, CheckAlign 2.0 computes two variations of the Shannon algorithm. For constructing sequence logos from the input of gapped and ungapped protein and DNA multiple alignments, CheckAlign 2.0 computes the canonical algorithm employed in CheckAlign 1.0, which has been described in depth ([5] and references therein). For accounts of both the abundance and evenness of the species present in a community sample, CheckAlign 2.0 computes the Shannon-Weaver diversity index (H) and the Equitability (EH) according to the following algorithms, respectively.
Algorithm 1Algorithm 2
Here, S is the total number of species in the community (richness) and pi is the proportion of S made up by the i-th species. Also, HMAX = ln S (equitability assumes a value between 0 and 1, where 1 indicates complete evenness).
CheckAlign 2.0 is available as an online server and as standalone software, shown in Figure 1 and Figure 2, respectively. The online server version has been programmed in PHP [URL 3] and JavaScript [URL 4,5] and is divided into two sections; the first gives access to the logo maker that builds sequence logos using multiple alignment inputs in FASTA format. The logo section of the PHP server includes an implementation of HMMER [URL 6] for users interested in creating a hidden Markov model (HMM) profile [4] based on the alignment in analysis. The second section allows the user to obtain diversity index estimates as described above from one or more community samples and with different numbers of species. The standalone software has been implemented in the Java programming language [URL 7] and has been completely rewritten using the SWT libraries to achieve a look and feel closer to the native operating system in which it is running. It shows similar organization and functions to those of the web server, allowing the user to create logos and make diversity estimates using his own computer (the software does not include the HMMER implementation, which is only available on the web version). Both standalone and online versions can now export logos not only to Postscript but to PDF files as well.
Installation
The CheckAlign 2.0 standalone application is distributed as an installer for Windows XP/Vista/7 (32 bit and 64 bit), a self-extracting disk image for Mac OS X 10.5 or later (64 bit), and a compressed tarball archive for Linux 2.6 kernel series or later (32 bit and 64 bit).
Requirements
The standalone application requires Java 6 or later. The minimum system requirements for this software are a PC with a Pentium 4 1.5 GHz or AMD Athlon XP 1500+ processor or higher with at least 1 GB of RAM.
Acknowledgments
The development of CheckAlign 2.0 has been partly supported by Grant IDI-20100007 from CDTI (Centro de Desarrollo Tecnológico Industrial) and by Torres-Quevedo Grants PTQ-09-01-00020 and PTQ-09-01-00670 from MICINN (Ministerio de Ciencia e Innovación) in Spain.
Funding to pay the Open Access publication charges for this article was provided by the University of Valencia
License and distribution
CheckAlign 2.0 is owned by Biotech Vana S.L. and is freely available as both an online server version and as a standalone application at [URL1, and URL 2]. The software and its source code are distributed under the terms of the Eclipse Public License v1.0 [URL 8] (formerly Common Public License 1.0) considered in the agreement for open source applications that you should accept during the installation of this tool.
Reference List
- Shannon CE, Weaver W: The mathematical theory of communication. University of Illinois Press; 1963.
- Schneider TD, Stephens RM: Sequence Logos - A New Way to Display Consensus Sequences. Nucleic Acids Research 1990, 18: 6097-6100.
- Schneider TD, Stormo GD, Gold L, Ehrenfeucht A: Information content of binding sites on nucleotide sequences. J Mol Biol 1986, 188: 415-431.
- Eddy SR: Profile hidden Markov models. Bioinformatics 1998, 14: 755-763.
- Llorens C, Futami R, Vicente-Ripolles M, Moya A: The CheckAlign logo-maker application in analyses of both gapped and ungapped DNA and protein alignments. In Biotechvana Bioinformatics. Biotechvana, Valencia; 2008:SOFT: CheckAlign.
URLs
- Checkalign 2.0 Server: http://gydb.org/tools/checkalign
- Checkalign 2.0 software: https://biotechvana.com/checkalign/index
- PHP programming language: http://php.net
- Client-side JavaScript reference: http://docs.sun.com/source/816-6408-10/contents.htm
- ECMAScript language specification: http://www.ecma-international.org/publications/standards/Ecma-262.htm
- HMMER: http://hmmer.janelia.org
- Sun Microsystems: http://www.java.com
- Eclipse Public License - v 1.0: http://www.opensource.org/licenses/eclipse-1.0.php





{kind=link}
{kind=link}
{kind=link}
{kind=link}