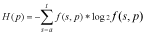
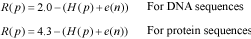
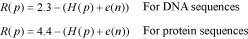
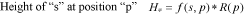
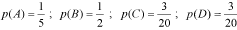
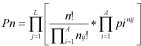
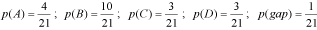
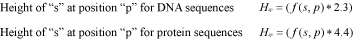



Abstract
In this paper we introduce CheckAlign, a logo-maker application that allows users to create consensus graphical representations from the input of both gapped and ungapped alignments using information theory. The application also allows users to generate a logo using a relative-frequency algorithm to overestimate the true consensus of distantly related sequences when conventional algorithms fail. CheckAlign is available as a free online server written in PHP, and as a Java application. This means that the tool runs on most personal computers (PCs) as a standalone program.
Availability
Available online February 1, 2008. ChekAlign is distributed as a server version written in PHP language and as a standalone software. CheckAlign server is freely accessible at http://gydb.uv.es/servers/checkAlign/checkalign_form.php. The Java application and source code are distributed under the terms of the Common Public License (URL 1) considered in the agreement for software and database tools provided in open source (URL 2).
Introduction
The construction of consensus sequences aids in the identification of conserved motifs that may be characteristic of protein domains. Consensus sequences are especially useful to classify proteins into families, and in order to assign putative physiological roles to proteins (1). Sequence logo methodology consists in the creation of graphical representations of the general consensus of DNA or protein multiple alignments. In every position, each residue is a letter whose height is proportional to its frequency per position multiplied by the information content of each position measured in bits (2). Letters are placed such that the most frequent one is positioned at the top, and the methodology is especially useful to decipher the order of predominance and relative frequencies of residues at every position, providing a significant view of the patterns that characterize the architecture of a gene or a protein. Nevertheless, protein and DNA alignments are not always well preserved in terms of similarity. In many cases, alignments demand manual refinement and introduction of gaps, for this reason a number of families of distantly related proteins do not have enough information content to build a significant logo. With the aim of constructing a versatile logo-maker application useful in the analysis of both conserved and non-conserved proteins we have designed CheckAlign.
Overview
Formats
The server version (Figure 1 left) builds the logo in two possible formats - PNG and PostScript - and additionally runs HMMER (URL 3) for users interested in creating a Hidden Markov Model (HMM) profile (3) based on the alignment analysis; the standalone JAVA version (Figure 1 right) allows users to build the logo in PostScript format.
Functions
CheckAlign is an application inspired in the information theory approaches of Schneider et al. (for more on this topic see (2,4-6)). CheckAlign provides two options for creating logos, “Shannon's algorithm” and “Relative frequency algorithm”. The tool reads the input of a multiple alignment in FASTA format and constructs a logo of the general consensus of this alignment. In the case of DNA logos, purines (a, g) are represented in black and pirimidines (c, t) in red. In protein representations, basic residues (K, H, R) are represented in red, hydrophobic residues (A, L, I, V, M, Y, F, W) in black, amino acids frequent in Β-turns (G, P) in grey, nucleophile amino acids (S, T) in violet, acidic residues (D,E) in orange, relatives amides (N,Q) in green, and cysteine (C) in blue.
Methodology
Shannon's algorithm.Following information theory principles (2, 4) the first option constructs a logo using Shannon's algorithm and considers the uncertainty measure as:
Algorithm 1H(p) is the uncertainty at position p, and s is one of the nucleotide (a, c, g or t) or amino acid (L, I, V, M, A, G, P, F, W, Y, H, K, R, D, N, E, Q, T, S or C) species. Note that f(s, p) is the frequency of the nucleotide or amino acid species s at position p. With this option users may build the logo from an ungapped alignment using the conventional methodology summarized in (2, 4). Here, the maximum uncertainty by position in a multiple alignment is log24 = 2 for DNA alignments, and log220 = 4.3 for protein alignments. The amount of information (R) by position is
Algorithm 2e(n) is the correction factor required for small samples of sequences.
Alternatively, users may build the logo from a gapped alignment considering the gap as another nucleotide or amino acid species. Here, instead of 2 and 4.3, CheckAlign considers the maximum uncertainty by position to be log25 = 2.3 for DNA sequences, and log221 = 4.4 for protein sequences, R is thus:
Algorithm 3In both cases, the size of s printed in the logo is determined by multiplying its frequency by R at position p within a multiple alignment:
Algorithm 4The application also allows users to build the logo using the correction factor (recommended for small alignments) and depicts standard deviation bars. With slight variations, the indications of the Appendix in Schneider et al. (4) were followed to program four PHP-scripts with which we have obtained four tables of statistics for sampling uncertainty and variance. These tables are available in the CheckAlign folder generated when installing the application and the PHP scripts can be downloaded from the CheckAlign site at Biotechvana Bioinformatics (see section “Installation” below). Two of these tables apply for DNA alignments; one considers equiprobable alignment composition for four nucleotide species (a, g, c, t) in ungapped DNA alignments; the other table assumes equiprobable composition considering the gap as a fifth nucleotide species in gapped alignments. The other two apply for protein alignments, one considers ungapped alignments and contemplates four groups of similarity A=(D, E, N, Q);? B=(G, P, A, L, I, V, M, Y, F, W); C= (R, H, K); D=(T, S, C). Here, while each amino acid has the particular probability of 1/20 in the alignment composition each physico-chemical group has been assigned the following probability:
Algorithm 5?The amino acid diversity within the protein multiple alignments is thus reduced to only four physico-chemical species; basic, acidic and relatives, hydrophobic, and nucleophilic. This facilitates the calculation of all possible combinations of ?n? nucleotide or amino acid species in a multinomial distribution required to infer the correction factor.
Algorithm 6The second table applies to gapped alignments and considers an additional group provided by the gap. Probabilities in the alignment composition are thus:
Algorithm 7Relative frequency algorithm. The second option for building logos with CheckAlign is simple but must be considered carefully because it is only implemented to facilitate the analysis of distantly related sequences when conventional algorithms fail in constructing the logo. This option always reports a logo but the representation results in an overestimation of the true consensus (so please read “Empirical example” section below). With this option, CheckAlign constructs the logo using a na?ve relative frequency algorithm multiplied by the maximum uncertainty to assign proportional height to residues.????
Algorithm 8This option allows users to decide the minimum frequency (in percentage) to be printed in the logos (by default 10%) and eliminates from the logo those regions displaying no information content.
Empirical example
In Figure 2a we show a logo representation constructed from the alignment of ninety primer binding site DNA motifs using the relative frequency option (notice the overestimation of information content in this logo). In contrast, when constructing the logo using Shannon's algorithm (Figure 2b) the representation offers a more informative perspective of which nucleotides are relevant in the primer binding site motif. Despite this, the information content overestimation caused by the relative-frequency algorithm may be advantageous in many cases where, due to high divergence, Shannon and other algorithms fail in constructing an informative logo. An example is clan AA: we used CheckAlign to determine a preliminary wide-range consensus for clan AA, a supergroup of proteolytic enzymes that groups a number of LTR retroelement-like and nonviral aspartic peptidases through less than 20% of identity (Llorens, C. and Moya, A., manuscript submitted for publication). Due to the absence of information content the tool failed (as also did other logo-making servers) in constructing an informative logo based on a single multiple alignment of 323 non redundant peptidase sequences, as shown in Figure 3a. In contrast the relative frequency algorithm was capable of resolving a rudimentary logo by taking advantage of overestimation of true consensus (Figure 3b). This logo is not significant under information theory principles, but disclosed six amino acid patterns which we call DTG/ILG templates. We used that template as a primary elucidation to explore the relationships of diversity within clan AA through the reconstruction of a clan AA Reference Database (CAARD), available at URL 4 within the Gypsy Database Project (7).
Installation
The software version of this tool is distributed in two versions: a self-installable executable package for Microsoft Windows platforms and a Java package (jar) compatible with all platforms. Java applications do not require to be installed on the computer to run as its source code is interpreted by the Java Runtime Environment previously installed on the computer. However, we provide a Windows installer which automatically creates shortcuts to the application. For executing the Windows installer version, simply double-click on the installer and follow instructions during installation. This process automatically generates desktop and start menu shortcuts. To execute the Java version of this software, open a command-line interface; locate the application folder named 'checkalign'; and finally, type 'java ?jar checkalign.jar'. To open a command line interface in Windows systems press the taskbar's 'Start' button; select 'Run...'; type 'cmd.exe' and accept. The server version is available as a PHP script and requires an HTTP web server (see section “Requirements” below). To run the calculation table (the PHP scripts) first, make sure that a web server engine and a PHP application server are properly installed on your system (see section “Requirements” below). Once the web server engine and a PHP server are properly installed and working, unpack the package containing this script on your server's public folder which is specified in its documentation. A folder named 'calhn' will be created. To access the calculation table utility, open a web browser and type the following URL location: http://localhost/calhn/index.htm.
Requirements
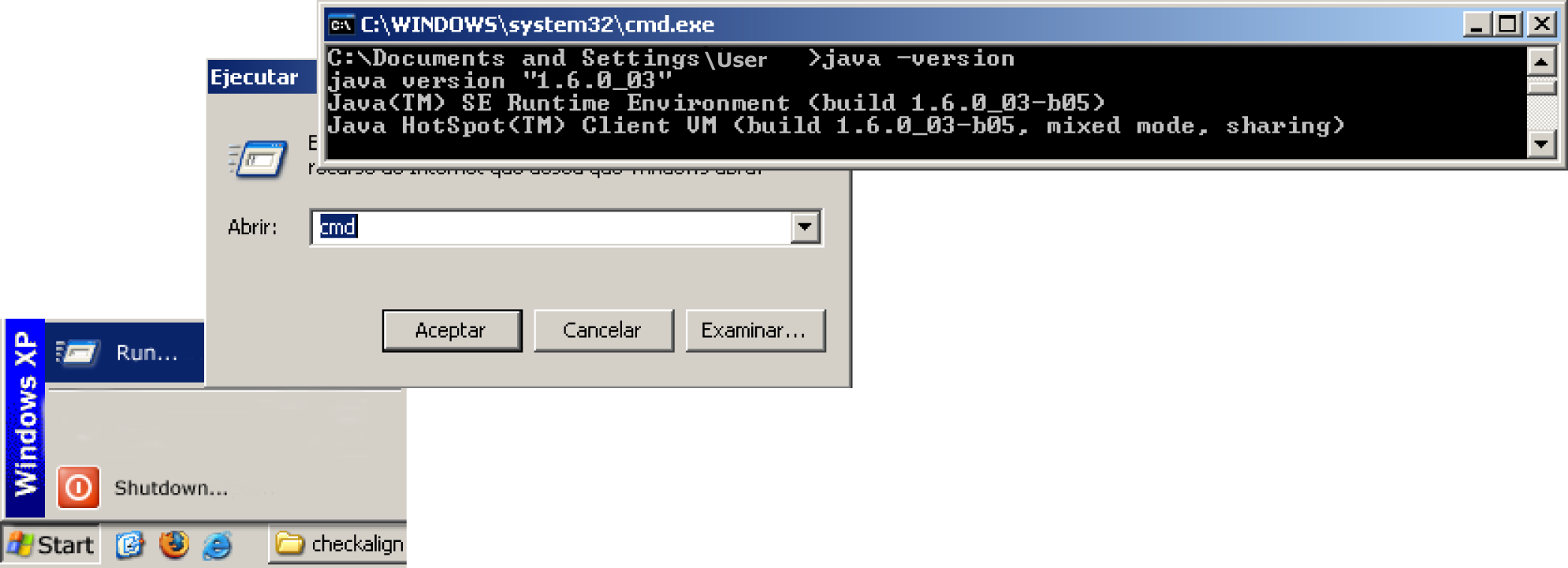
The software version has been designed as an open source Java application and therefore runs on most PCs as a standalone program. Since CheckAlign is a Java application, make sure before installing the tool that a Java Runtime Environment (JRE) is previously installed on your computer. JRE is a software bundle from Sun Microsystems that allows a computer system to run Java applications. A JRE can be freely downloaded and installed from Sun Microsystems' Web site at URL 5. This application has been tested in a JRE version 1.4. To know if a JRE is currently installed on a Microsoft Windows operating system, click the taskbar?s “Start” button; select “Run”; type “cmd.exe” to open a command-line window;?and finally, type "java -version" to know the current version installed on your computer, as shown in Figure 4. If an error message is prompted, it means that JRE is not properly installed on your computer.
To process the PHP scripts you will need a PHP application server and a web server engine. A web server engine is a computer program responsible for accepting HTTP requests from user's web browsers and returning HTML web pages, images and other files. An application server is software that helps a web server to process web pages containing server-side scripts that cannot be processed by a regular web server engine. When a dynamic page is requested by a visitor's browser, the web server calls the application server for processing scripts before sending the page to the browser. You can download and install a web server like Apache from URL 6 for Windows and Linux platforms or an IIS (Internet Information Services) web server for Windows platforms which comes included in Windows server distributions. Then, install the PHP application server which can be downloaded at URL 7. Instructions on installation are provided in its corresponding web sites.
Concluding Remarks
CheckAlign is an easy-to-use application that allows users to build logo representations online and on PCs, using the conventional methodology introduced by Schneider and Stephens or using the na?ve relative frequency approximation. This last option does not offer direct significant results; nonetheless it is helpful to establish a preliminary visualization of a consensus sequence and offers an alternative insight of how to approach further analyses in those cases where conventional methodology fails to construct a logo.
Acknowledgments
We thank Rachel Epstein for language revision and the Servei Central de Suport a la Investigaci? Experimental (SCSIE) (URL 8) at UVEG for technical support. Biotechvana Bioinformatics has been awarded the NOVA 2006 by IMPIVA and Conselleria d'Empresa, Universitat I C?encia of Valencia. The research has been partly supported by grants IMCBTA/2005/45, IMIDTD/2006/158 and IMIDTD/2007/33 from IMPIVA, and by grant BFU2005-00503 from MEC to AM.
Literature
- 1. Schug,J., Diskin,S., Mazzarelli,J., Brunk,B.P. and Stoeckert,C.J., Jr. (2002) Genome Res., 12, 648-655.
- 2. Schneider,T.D. and Stephens,R.M. (1990)Nucleic Acids Research, 18, 6097-6100.
- 3. Eddy,S.R. (1998)Bioinformatics, 14, 755-763.
- 4. Schneider,T.D., Stormo,G.D., Gold,L. and Ehrenfeucht,A. (1986)J. Mol. Biol., 188, 415-431.
- 5. Shannon,C.E. (1997) MD Comput., 14, 306-317.
- 6. Pierce,J.R. (1980) Dover Publications,Inc., New York.
- 7. Llorens,C., Futami,R., Bezemer,D. and and Moya,A. (2008) Nucleic Acids Research (NAR) 36 Database-Issue):38-46
URLs
- 1. CheckAlign Server: http://gydb.org/index.php/Checkalign_sequence_logos
- 2. Common Public License: http://www.opensource.org/licenses/cpl1.0.php
- 3. Biotechvana agreement: http://biotechvana.com/loader.php?section=contents&page=terms_ocl
- 4. HMMER: http://hmmer.janelia.org
- 5. Clan AA Reference Database: http://gydb.uv.es/index.php/Phylogeny:CAARD
- 6. Sun Microsystems: http://www.java.com
- 7. Apache Server: http://www.apache.org
- 8. PHP Programming Language: http://php.net
- 9. SCSIE, Universitat de València: http://scsie.uv.es





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}